THE PROBLEM
Understanding protein-ligand recognition is crucial in drugs discovery.
Most of the features used for describing such interactions are parametric.
This practically means that, if there are not suitable parameters for a particular atom, functional group or interaction, either the calculation fails or a rough approximation is provided.
We wanted to know whether or not non-parametric chemical descriptors could compete with parametric ones.
THE SOLUTION
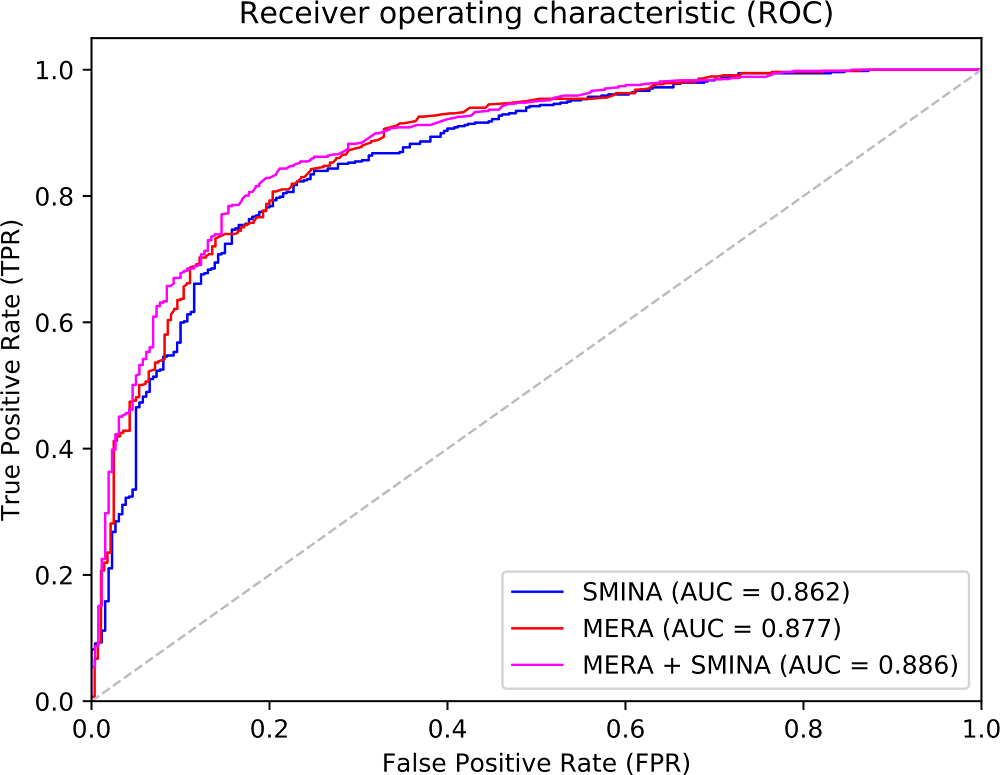
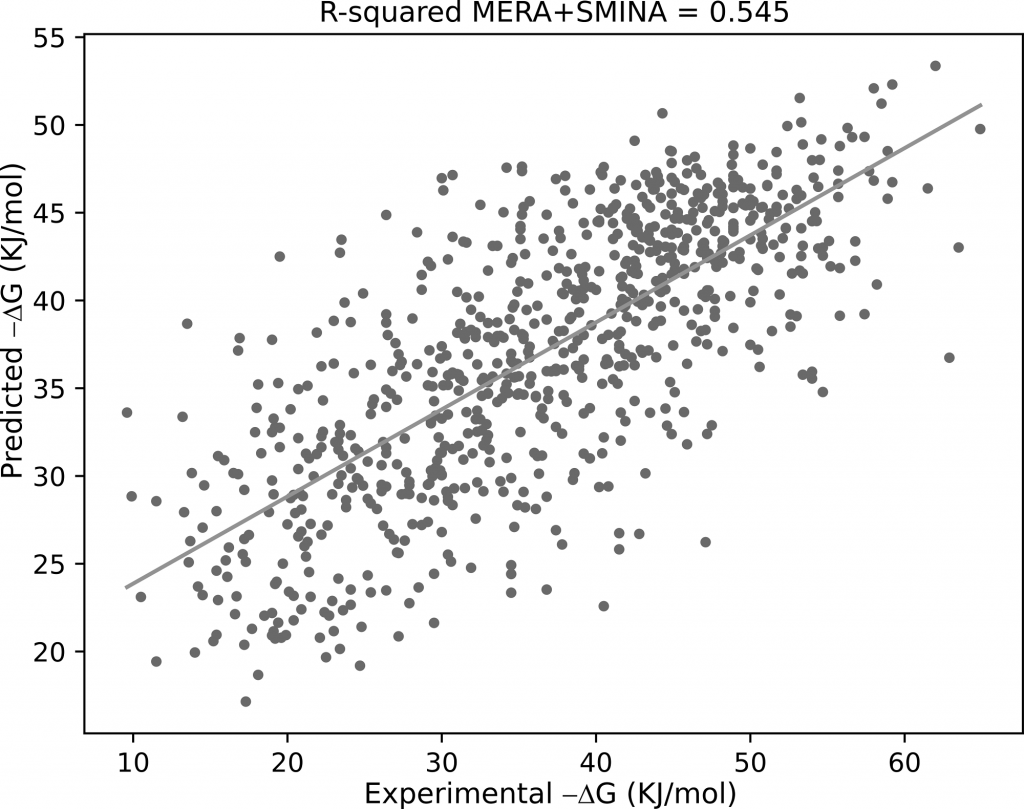
We tested two different non-parametric descriptors sets by using machine learning algorithms.
One of such sets provided better results than those obtained with state-of-the-art parametric sets.
A Python 3 / Bash automatic workflow was developed that:
- Downloads and fixes the PDB files.
- Minimises the protein-ligand binding site.
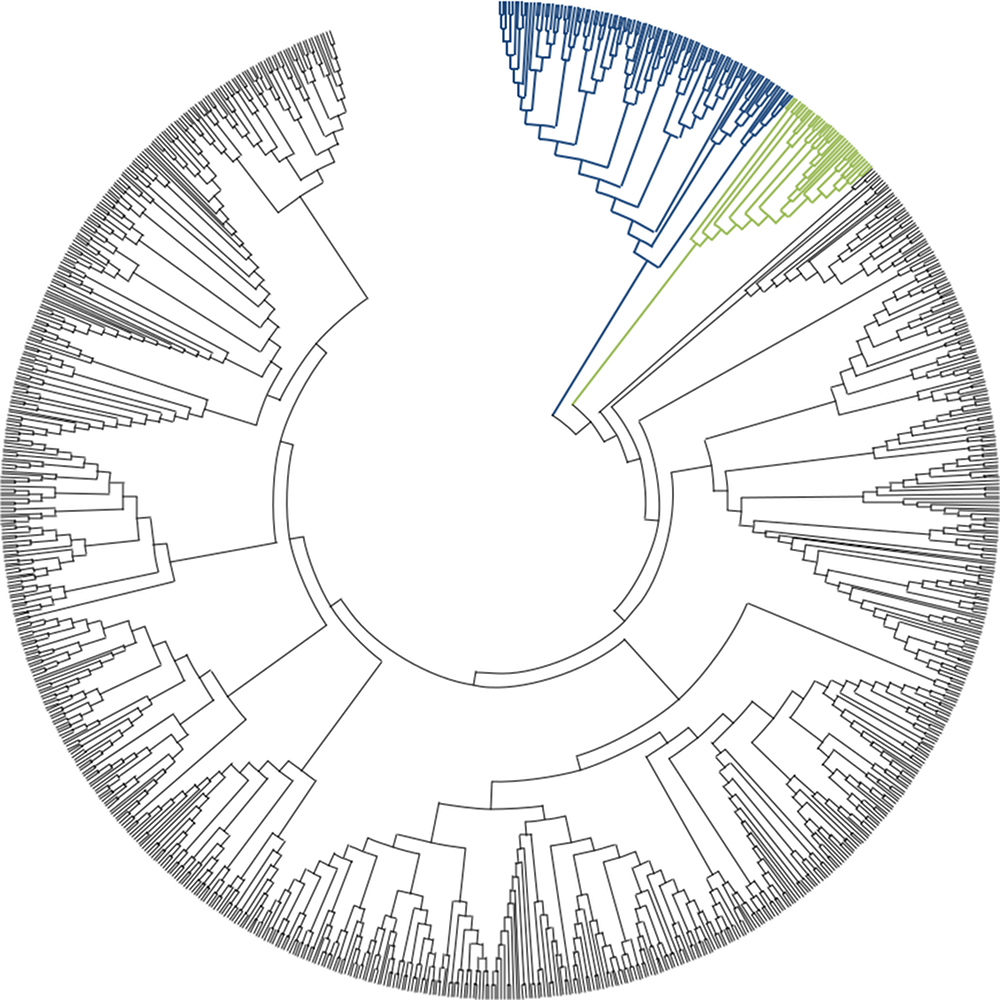
- Splits the testing, validation and training sets according to different criteria.
- Trains machine learning models.
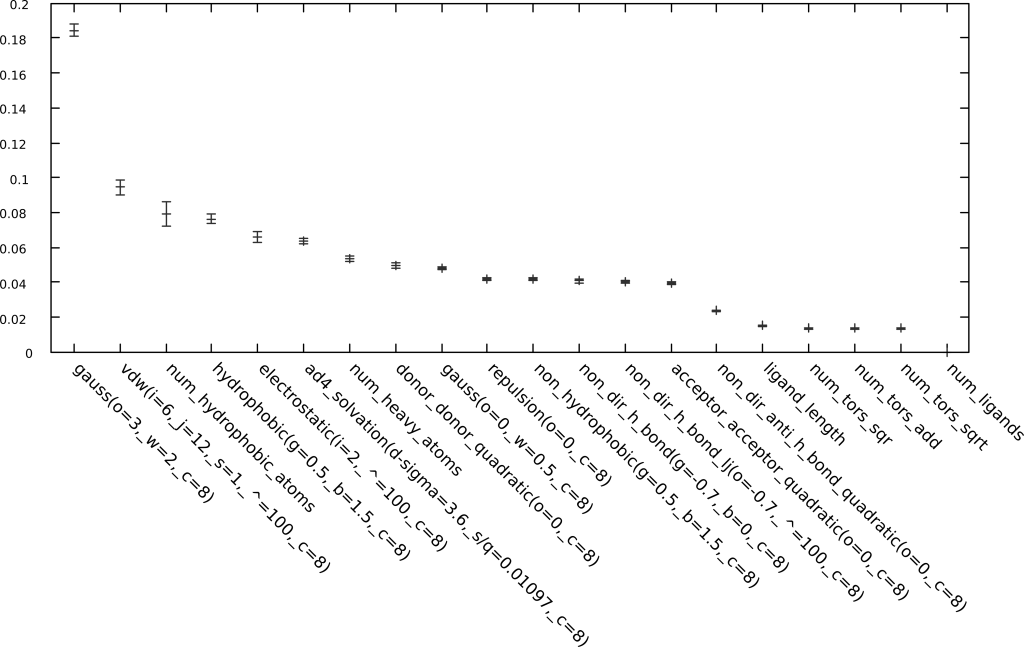
- Determines which, among thousands of descriptors, contribute more to model accuracy.
- Plots the results.
THE OUTCOME
The results have been published in a peer-reviewed scientific journal.